
FIBRODISPLASIA OSSIFICANTE PROGRESSIVA: UMA REVISÃO BIBLIOGRÁFICA
SOBRE O ALUNO
Jullia Paoli Bodenmüller
Larissa Bado
Marcelo Rocha Soares da Silva
ARTIGO
INTRODUÇÃO
A FibrodisplasiaOssificante Progressiva (FOP) é uma doença genética rara caracterizada pela ossificação de tecidos conectivos,⁵ que atinge cerca de 1,3 a cada 1 milhão de pessoas e não distingue raças, gêneros e etnias.⁹
A FOP é uma doença congênita e apesar desta característica, só apresenta formação de ossos extras a partir do nascimento.⁴ Esse processo é conhecido como “ossificação heterotópica”.¹¹ Como um dos primeiros sinais pode-se perceber a má formação dos háluces, onde esses são encurtados e curvados para dentro (formato em valgo), presentes em 75-90% dos casos.¹⁰
Tem-se conhecimento de que os primeiros casos documentados correspondem ao ano de 1700, onde a doença era conhecida como “miositeossificante progressiva”, e apenas em 1970 é que a doença passou a ter o nome de FibrodisplasiaOssificanteProgressiva.³,⁴
Em 2006, após muitos anos de estudos, a equipe do Dr. Frederick Kaplan (Escola de Medicina da Universidade da Pensilvânia) descobriu o gene causador da doença,¹² chamado de ACVR1, que está localizado no cromossomo 2. Devido a mutação do gene, ocorre uma desregularização da produção de Proteína Morfogenética Óssea (BMP), onde há então a calcificação dos tecidos conectivos.⁶
Os surtos, ou como são chamados “flare-ups”, são os sintomas da FOP em atividade. Quando se inicia surge uma inflamação, edemas dos tecidos, gerando assim a calcificação dos tecidos conectivos, dores e desconforto ao paciente. Deste modo há uma perda significativa de movimentos, como a constrição de mobilidade da caixa torácica, articulação temporomandibular e membros.³
As consequências da FOP são cumulativas, levando o paciente a enfrentar diversas limitações que implicam nos âmbitos psicossocial, físico, mental e emocional.⁸
Apesar de existirem estudos clínicos, a FOP ainda não tem cura. Com isso, os pacientes têm a necessidade de estar utilizando medicamentos como corticoides e anti-inflamatórios não esteroides para alívio dos sintomas.³,⁸
O presente trabalho tem por objetivo analisar os critérios de diagnóstico, fisiopatologia, sinais e sintomas e tratamentos da FibrodisplasiaOssificante Progressiva (FOP), baseados nas informações que tem-se atualmente sobre a doença. A metodologia utilizada será de levantamento bibliográfico.
PALAVRAS CHAVE:FibrodisplasiaOssificante Progressiva; Complicações; Tratamento.
FIBRODISPLASIA OSSIFICANTE PROGRESSIVA-(FOP) FibrodisplasiaOssificante Progressiva (FOP) é uma doença genética congênita rara na qual um esqueleto extra é formado. A doença caracteriza-se pela ossificação de tecidos conectivos como ligamentos, tendões, músculos, fáscias, aponeuroses, entre outros. Esse processo recebe o nome de “ossificação heterotópica”.¹³ Em geral, os músculos do coração, diafragma, dos olhos, da língua e músculos lisos viscerais são poupados dessa condição.¹
A doença não difere raças, gêneros e etnias, sua incidência é de 1,3 por milhão de pessoas.⁹ Atualmente, cadastrados no InternationalFibrodysplasiaOssificans Progressiva Association (IFOPA), existem 1072 casos no mundo, com uma maior prevalência na América do Norte (231 casos), na Europa (198 casos) e na América Latina (161 casos). 60% da população mundial afetada pela FOP está entre os 29 anos, sendo que a média de vida dos portadores circula entre 40-45 anos de idade.⁵,⁸ Esses dados sugerem que a incidência da FOP pode ser maior que a teoria, uma vez que há pacientes em fase de diagnósticos e pacientes não cadastrados no IFOPA.⁵
Em 1692, foi registrado o primeiro relato da doença, onde Guy Patin intitulou a paciente como “mulher que virou madeira”. Só em 1918, Rosensum realizou uma revisão médica literária sobre a FOP, revelando 115 casos.¹,¹⁷ Em suas primeiras décadas, a doença era conhecida como “miositeossificante progressiva”, considerando que apenas os músculos sofriam o processo de inflamação, tornando-se posteriormente um pedaço de osso.³,⁴ Já no ano de 1970, através dos estudos do Dr. Victor McKusick, a doença passou a ter o nome de FibrodisplasiaOssificante Progressiva, reconhecendo-se que não apenas músculos se consolidavam, mas também outros tecidos conjuntivos.³,⁴ No ano de 2006 ocorreu a descoberta do gene causador da doença, dando-se um grande passo na obtenção de informações para um possível tratamento.²⁰
“A FOP era, até recentemente, um dos mistérios mais ilusórios da medicina. Para os pacientes que sofrem de FOP, é uma dolorosa metamorfose para a imobilidade progressiva e um obstáculo ao longo da vida para a liberdade física. Embora tratamentos definitivos e curas ainda não estejam disponíveis, os objetivos da pesquisa da FOP são bem articulados: estabelecer as bases genética, molecular e celular da FOP; e usar esse conhecimento para estabelecer prevenção, tratamento e, eventualmente, cura.”¹⁹
QUEM FOI HARRY RAYMOND EASTLACK?
Harry Eastlack, nascido na Filadélfia em novembro de 1933, tornou-se famoso após doar seu corpo e registros médicos para o The MutterMuseumoftheCollegeofPhysiciansof Philadelphia. Durante a sua fase adulta, tomou essa decisão com o objetivo de auxiliar nos estudos a fim de conhecimento da FOP.²²
Seu esqueleto é o único totalmente articulado no mundo, onde é visível as consequências deixadas pela doença durante seus anos de vida. Calcificações em todo o corpo decorrentes da FOP tornam o esqueleto de Harry quase uma peça contínua.²¹
Em novembro de 1973, faleceu devido a uma pneumonia:
“revelando uma janela para os mistérios médicos e desafios científicos da FOP, e exemplificando a dura realidade da doença mais do que qualquer gráfico, slide ou descrição clínica poderia realizar”.²¹
Seu esqueleto encontra-se até hoje em exposição no The MutterMuseum.⁵,²¹
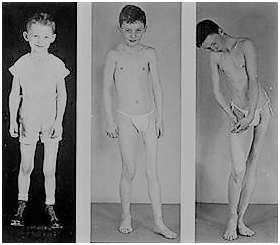
Fig. 1: Harry Raymond Eastlack aos 6 anos, 9 anos e 12 anos, respectivamente.¹⁵
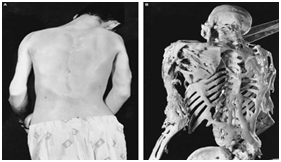
Fig. 2: Harry Raymond Eastlack aos 40 anos e seu esqueleto no The Mutter Museum.²¹
FISIOPATOLOGIA
A FOP é uma doença genética e está diretamente ligada ao DNA, que é responsável por repassar informações necessárias para o perfeito desenvolvimento do nosso corpo, bem como tudo o que acontece nele. Cada DNA possui 23 pares de cromossomos, que são divididos em pequenos genes. São compostos por quatro letras que codificam o código genético. Uma vez que estas letras não estão na ordem combinada, há um desequilíbrio na homeostasia. Esta desregularização acontece na FibrodisplasiaOssificante Progressiva.³ Uma mutação ocorre no gene ACVR1, onde uma letra em 6 bilhões é substituída, alterando todo o código genético.³ Estudos apontam que, em 97% dos casos, a mutação se dá na sequência R206H.⁹
O portador da FOP possui uma cópia do gene normal e outra cópia defeituosa. O gene está localizado no cromossomo 2 e codifica o receptor ACVR1/ALK2, que está ligado a Proteína Morfogenética Óssea (BMP) e que regula o desenvolvimento e o crescimento de tecido ósseo.⁶
Pacientes acometidos pela doença possuem uma produção de Proteína Morfogenética Óssea 4 (BMP4) em excesso e pequenas produções de proteínas antagonistas das BMPs, como Noggin e Gremlin.²³ Em pacientes não portadores existe um equilíbrio na produção dessas proteínas. Enquanto a BMP4 estimula a produção óssea, as proteínas antagonistas tem como função inibi-la quando produzida em excesso, desse modo controlando a produção óssea. No entanto devido ao desequilíbrio na produção dessas proteínas, os pacientes com FibrodisplasiaOssificante Progressiva acabam tendo excesso de produção óssea e falta de inibidores, que acabam resultando nas calcificações dos tecidos conjuntivos.¹,²
A BMP4 é sintetizada pela musculatura esquelética e pode ser produzida nos locais onde houve traumatismos. Devido a diminuição da produção de proteínas antagonistas em paciente com FOP, essas BMPs produzidas nos traumas acabam ficando nos tecidos, desencadeando assim os sintomas da FOP.²
DIAGNÓSTICO E QUADRO CLÍNICO DO PACIENTE
A FOP geralmente apresenta um padrão de progressão: axial-apendicular, cranial-caudal e proximal-distal. Os pacientes acometidos pela doença são reconhecidos pela formação de dois esqueletos: um formado durante a embriogênese (esqueleto normotópico) e um outro que se desenvolve após o nascimento (esqueleto heterotópico).⁴
Os primeiros sinais e sintomas costumam aparecer na primeira década de vida, iniciando-se em média aos 3 anos de idade.¹⁷ As calcificações iniciais tendem a se manifestar na coluna vertebral e nas articulações proximais dos membros superiores e inferiores.¹¹
Apresenta como particularidade a malformação dos háluces, onde esses se encontram encurtados e curvados para dentro (formato em valgo),¹⁴,⁷ e as cabeças de fêmur podem ser largas e encurtadas. Há uma grande chance de desenvolvimento de osteocondromas.¹
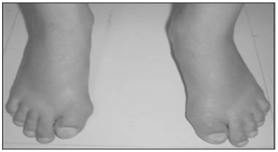
Fig. 3: Malformação dos háluces.²⁴
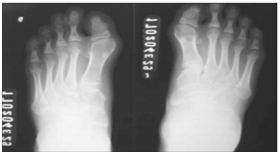
Fig. 4: Radiografia AP de pé, onde observa-se os háluces encurtados e em valgo.²⁴
Seus sintomas aparecem através dos flare-ups, ou surtos, que é o momento da FOP em atividade, podendo durar semanas ou até mesmo meses. Não se sabe ao certo como surgem, mas uma vez iniciado surge o processo de inflamação. Nesse processo podem aparecer nódulos, edemas e a área pode ficar quente e avermelhada, além do gradativo enrijecimento das partes afetadas. Algumas pessoas relatam ter apresentado febre baixa e dores nas regiões afetadas também.³ Em muitos casos, o surgimento destes nódulos podem ser facilmente confundidos com tumores. Isso acaba acarretando em um diagnóstico errôneo, submetendo o paciente a procedimentos considerados perigosos aos portadores da FOP, como por exemplo cirurgias e biópsias. Por isso a importância de ter-se um diagnóstico preciso e não invasivo.¹¹
Esses surtos podem, ou não, preceder um crescimento ósseo.³,¹⁰ Além dos surtos, outros fatores podem acarretar uma calcificação. Injeções intramusculares, cirurgias, simples procedimentos odontológicos e até mesmo quedas podem fazer com que um novo pedaço de osso cresça onde não deve.³,⁴ A partir do momento que o portador assume uma postura única e limitada, por conta do enrijecimento, pode-se denominar síndrome de “Stone Man”.⁷,¹⁶
Apesar dos ossos extras nos portadores da FOP, o índice de fraturas não é maior do que em pessoas que não possuem a doença. Porém a consolidação dessas fraturas será mais rápida, exigindo menor necessidade de procedimentos cirúrgicos e imobilizações com gesso. Vale ressaltar que os ossos heterotópicos podem ser fraturados e consolidados como um osso normotópico.³,⁸
Calcificações heterotópicas precoces, que por muitas vezes não são detectadas por imagens radiográficas comuns, são facilmente sinalizadas através da cintilografia óssea, um exame altamente sensível.¹⁷ É injetado no paciente um radiofármacochamado Metilenodisfosfonato marcado com Tecnécio 99m, cujo mesmo é absorvido pelas estruturas ósseas de acordo com a função osteometabólica e através do equipamento Gama-Câmara as calcificações são detectadas.¹⁸,²⁹
Exames de imagens, como Raio-x, Tomografia Computadorizada e Ressonância Magnética são meios complementares para diagnóstico da FOP quando há calcificações nas lesões. Através dos testes de DNA do gene ACVR1 pode-se obter a confirmação do diagnóstico clínico.¹⁹
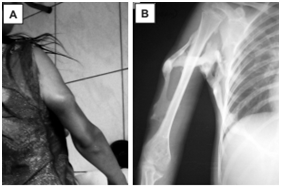
Fig. 8: Braço direito da paciente com ossificação heterotópica (A). Radiografia de úmero AP, onde visualiza-se ossificação em partes moles no úmero estendendo-se para escápula direita (B).⁸
PRINCIPAIS COMPLICAÇÕES DA FOP
Gravidez: Uma mulher com FOP possui a capacidade de engravidar, porém os riscos são extremos, tanto para a saúde da mãe quanto a do bebê. As limitações físicas causadas pelas ossificações heterotópicas em todo o tronco suprimem o crescimento e a adaptação do bebê no útero, comprometendo assim o período da gestação.³
Diversas complicações podem ocorrer em uma gravidez de uma paciente com FOP, sendo as principais: riscos de surtos, dificuldades respiratórias, sofrimento fetal, riscos durante o parto e a probabilidade de 50% da criança nascer com FOP.³
Articulação Temporomandibular: A articulação temporomandibular (ATM) é tipicamente afetada pela FOP. Estudos apontam que em 71% dos pacientes tem restrições na mandíbula antes dos 20 anos³ e que o músculo comumente afetado é o masseter.²⁶
Ao ocorrer a anquilose da ATM, ou seja, a fusão de superfícies que compõe a articulação, o portador pode ter dificuldade na mastigação, fala e higiene, além do comprometimento da abertura bucal.²⁶
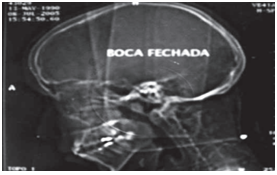
Fig. 5: Escanograma de Tomografia Computadorizada visualizando-se a mandíbula sem abertura bucal.²⁷
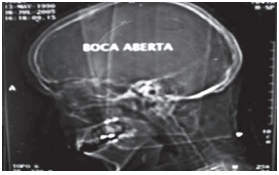
Fig. 6: Exame de Tomografia Computadorizada visualizando-se a diminuição da abertura bucal.²⁷
Em alguns casos da fusão extrema da mandíbula, a plástica do esmalte dentário pode ser recomendada. Um procedimento onde retira-se uma pequena camada do esmalte e permite uma maior abertura bucal. Antes desse processo, um exame de Raio-x deve ser realizado para visualizar-se a localização da polpa dos dentes.³
Sistema Cardiorrespiratório: A função cardiopulmonar anormal é uma das principais características da doença,²⁵ devido às más formações das articulações costovertebrais e anquiloses das mesmas; e calcificações dos músculos intercostais,paravertebrais e aponeuroses.¹⁹ Devido a estas restrições, os portadores da doença dependem da respiração diafragmática.³
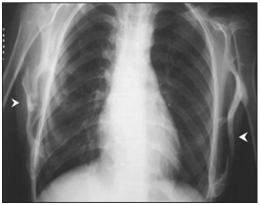
Fig. 7: Radiografia de tórax PA, com calcificações em tecidos moles na parede torácica.²⁴
Conforme a progressão da FOP, os ossos extras e a posição do corpo podem comprimir a caixa torácica, resultando assim na limitação dos movimentos respiratórios e desequilibrando o fluxo ventilatório. Estudos também demonstraram anormalidades eletrocardiográficas, indicando sobrecarga no lado direito do coração, cujo mesmo tem a função de bombear sangue para os pulmões.³
Pacientes com FOP avançada não devem fazer uso de oxigênio em ambiente não monitorados, pois há retenção crônica de dióxido de carbono maior que o normal.¹⁹
Portadores da FibrodisplasiaOssificante Progressiva possuem uma alta probabilidade de desenvolver complicações da gripe, resultando em infecções respiratórias como a pneumonia. A pneumonia é uma doença pneumocócica que afeta os pulmões e é uma das principais causas de mortalidade em pessoas com FOP. Uma vez que há restrição respiratória, estes pacientes tendem a ter uma dificuldade no combate a esta infecção.³
“Gargalhar também pode ajudar e é algo que qualquer pessoa pode fazer. Gargalhar exercita os músculos do diafragma, abdômen e pulmões, bem como os músculos da face, perna e costas. Em alguns aspectos é como fazer um exercício de aeróbica. Gargalhar leva à inspiração profunda que envia sangue rico em oxigênio e nutrientes para todo o corpo”.³
TRATAMENTO
Atualmente não existe um tratamento para a FOP. Tudo que se tem são remédios que ajudam a aliviar a dor e os edemas. O laboratório Clementia está em fase de testes com o medicamento Palovaroteno (atualmente na fase 3).⁶ Em setembro de 2018 está previsto para iniciar estudos do medicamento Rapamicina, na Universidade de Kyoto, que também promete ser um grande avanço para os tratamentos da FOP.¹⁵
Existem também medicamentos que ajudam no controle dos sintomas dos surtos com efeitos colaterais mínimos: corticosteroides, como a Prednisona, e anti-inflamatórios não esteroides, como os inibidores da cicloxigenase-2 (Cox-2).³
O uso da Prednisona deve ser iniciado logo após um surto, tornando-se assim útil na diminuição dos edemas e da inflamação. Porém o seu uso prolongado pode não ser benéfico epode até mesmo acelerar o processo de crescimento ósseo.³
Outra alternativa para a Prednisona, são as drogas anti-inflamatórias não esteroides ou inibidores da Cox-2, como o Ibuprofeno. Estas drogas atuam diretamente nas prostaglandinas, que são substâncias que causam a inflamação e contribuem para novas formações ósseas, diminuindo assim a atuação dessa substância e evitando uma ossificação heterotópica. Estudos demonstram que os inibidores da Cox-2 também tem propriedades anti-angiogênicas, ajudando assim a não alimentar o crescimento de um novo osso através dos vasos sanguíneos.³
Lembrando que todo e qualquer uso de medicação deve ser controlado por um médico responsável, levando-se em conta os prós e contras da droga em questão.
PALOVAROTENO E O ESTUDO MOVE
O Palovaroteno é um fármaco, administrado via oral, que está em fase experimental pelo laboratório Clementia. Tem como objetivo ser uma alternativa de tratamento para a FibrodisplasiaOssificante Progressiva.¹⁵ Segundo os estudos, esse medicamento é capaz de inibir a atuação da proteína morfogenética óssea (BMP), que é responsável pela formação óssea.⁶
Estudos apresentaram que o medicamento impediu as ossificações heterotópicas, tanto induzidas quanto espontâneas, e normalizou o crescimento do esqueleto, proporcionando a mobilidade. Cerca de 800 voluntários saudáveis e com FOP participaram do estudo.¹⁵
Atualmente o laboratório Clementia está realizando o estudo MOVE. É um estudo clínico confirmatório e experimental do uso do Palovaroteno no tratamento da doença.⁶
Os portadores da FOP possuem uma mutação no receptor da via da BMP, causando um aumento na sua atividade e enviando sinais para a mesma realizar a ossificação heterotópica. Estudos apontam que o Palovaroteno, inibe esses sinais enviados, evitando assim a formação de um novo osso.⁶
Observou-se em uma análise de dados que pacientes tratados, recebendo diariamente a medicação e tendo sua dose aumentada nos períodos de manifestação da doença, tiveram uma diminuição de 97% em novas ossificaçõesheterotópicas. A fase 3 do teste poderá confirmar esses dados.¹⁵ Em contrapartida, efeitos colaterais envolvendo a pele e mucosas foram relatados: pele e boca seca, coceira, vermelhidão e erupções cutâneas. Conforme a dose foi aumentada, os efeitos colaterais também aumentaram.⁶
O estudo clínico MOVE confirmatório de fase 3 conta com 80 portadores da doença, adultos e crianças acima de 4 anos. O objetivo é avaliar a redução de ossificação heterotópica com base na dosagem da medicação administrada diariamente e com aumento de dose no período de pico da doença, e também avaliar a segurança e a eficácia para futura aprovação global. Os pacientes irão ser acompanhados no tratamento durante dois anos.⁶
O Palovaroteno recebeu três designações de duas agências internacionais:
FastTrackandBreakthroughTherapy (terapia inovadora e urgente), da Agência de Administração de Alimentos e Medicamentos dos EUA (FoodandDrugsAdministration, FDA): indicando, com base nos dados clínicos, que o medicamento pode apresentar melhorias a doença e reconhecendo que ele pode ser benéfico ao tratamento.⁶
Medicamento Órfão, da Agência de Administração de Alimentos e Medicamentos dos EUA (FoodandDrugsAdministration, FDA) e da Agência de Medicamentos Européia (European Medicines Agency, EMA): reconhecendo que a FOP é uma doença rara e debilitante, onde há falta de tratamentos eficazes.⁶
Em abril de 2018, a comunidade da FOP Brasil recebeu a aprovação da Anvisa em relação a vinda dos estudos clínicos do Palovaroteno ao Brasil. Inicia-se agora a etapa de importação do medicamento, que pode ocorrer dentro de 60 dias ou mais.¹⁵
Stéphanie Hoffmann e Eric Soliman, da ClementiaPharmaceuticals, visitaram o Brasil para iniciar os preparativos para o ensaio clínico Palovaroteno e para implementar um projeto intitulado “Projeto Narrativo FOP”. O objetivo primário deste projeto é compreender como o sistema de saúde brasileiro atualmente apoia o diagnóstico e o atendimento de pacientes com FOP.¹⁵
OUTROS ENSAIOS CLÍNICOS
Pesquisadores, liderados por JunyaToguchida, criaram células IPS (células-tronco pluripotentes induzidas, que são células que voltam a se comportar como uma célula-tronco embrionária através de uma reprogramação no seu DNA²⁸) com as características da FOP e após adicionarem cerca de 6800 componentes nestas células, descobriu-se um agente imunossupressor capaz de prevenir a ossificação heterotópica. Esse agente é chamado de Rapamicina, medicamento que já é utilizado no tratamento de outras doenças.¹⁵
O comitê do Hospital da Universidade de Kyoto aprovou o ensaio clínico para que a segurança e eficácia da droga sejam testadas. Em setembro de 2018 está previsto para iniciarem-se os testes, com cerca de 20 pacientes tendo a idade superior a 6 anos, utilizando o fármaco Rapamicina.¹⁵
Outro ensaio clínico em atividade é com o medicamento experimental REGN2477, da Regeneron. O teste Regeneron LUMINA-1 está atualmente na Fase 2 e será testado pela primeira vez em pacientes com FOP. Tem como objetivo avaliar a segurança, a tolerabilidade e os efeitos do medicamento nas formações ósseas anormais em pacientes adultos com FOP.⁵
O estudo será feito com cerca de 40 pacientes com FOP. Metade receberá placebo durante 6 meses, seguidos de mais 6 meses de REGN2477, enquanto a outra metade receberá 12 meses seguidos do medicamento em questão.⁵
Ambos os ensaios são promissores, visando buscar mais de uma forma de tratamento para a doença, controlando as ossificações heterotópicas, melhorando a mobilidade e a vida dos pacientes acometidos pela FOP, aumentando assim a perspectiva de vida.
CONVIVENDO COM A FOP
Como já mencionado no decorrer deste artigo, o portador da FOP pode atingir um nível avançado de imobilidade, chamado síndrome de “Stone Man”.⁷,¹⁶
Essa condição irá forçar o paciente a adaptar-se das formas mais criativas possíveis, buscando conforto e condições para realizar atividades diárias.³
Camas ajustáveis, travesseiros com casca de trigo ou microesferas e almofadas ortopédicas são dicas para que o paciente fique totalmente apoiado e confortável dentro de suas limitações.³ Também há diversos tipos de cadeiras de rodas no mercado que oferecem uma forma segura de locomoção aos pacientes.³⁰
Adaptações criativas podem auxiliar muito na independência dos portadores da FOP: canos de PVC para estender objetos como escovas de cabelo, escovas de dente e talheres é uma alternativa de baixo custo e viável aos pacientes e suas famílias. Mãos mecânicas, sejam elas compradas ou ajustadas, auxiliam pacientes com calcificações em membros e tronco a pegarem objetos em diferentes alturas.³⁰
Por muitas vezes, os medicamentos oferecidos para a dor não são suficientes e alguns médicos acreditam que as emoções podem interferir diretamente na dor. Terapias como de relaxamento, treinamento de biofeedback, modificação de comportamento e gerenciamento de stress são algumas sugestões para o paciente lidar melhor com a dor.³
Melhorar as habilidades funcionais e gerenciar a dor de pessoas com FOP, podem melhorar a qualidade de vida permitindo-as realizar atividades que gostam de forma confortável.³
CONCLUSÃO
A FibrodisplasiaOssificante Progressiva é uma doença congênita rara, que foi descoberta há mais de 300 anos. Atualmente existem grandes estudos acerca da doença, sendo um assunto de interesse multidisciplinar. Porém o conhecimento geral ainda é muito escasso, levando a diagnósticos errôneos e submetendo os pacientes a procedimentos perigosos que acabam sendo um agravante.
Durante o decorrer da doença, diversas complicações podem acontecer oferecendo riscos ao portador. No estágio mais avançado, o paciente pode sofrer da síndrome de Stone Man.
Devido à falta de informações sobre a FOP, foi desenvolvido um cartão de emergência que consiste em informar sobre os principais aspectos da doença e os cuidados ao paciente. Assim, em qualquer emergência, será feito um atendimento correto, evitando o máximo de complicações a curto e a longo prazo.
No momento, ainda não existe uma cura. Diversos medicamentos estão disponíveis na tentativa de ajudar o paciente a suportar os sintomas, e também existem diversas opções criativas que podem auxiliar no dia a dia, possibilitando uma qualidade de vida ao portador da FOP.
REFERÊNCIAS
- ARAÚJO, E. M. V. M. ; OLIVEIRA, A. C. ; Fibrodisplasiaossificante progressiva: artigo de revisão. Revista de trabalhos acadêmicos Universo Recife, v.1, n.1. 2014.
- BARBOSA, B. F. ; SANTOS, L. C. S. ; MATTOS, A. M. H. ; QUEIROZ, C. S. ; Fibrodisplasiaossificante progressiva: relato de caso. Revista UNINGÁ, Maringá – PR, n.26, p. 103-109.2010.
- DELAI, P. L. R. ; KAPLAN, F. S. ; SHORE, E. M. ; O que é FOP? Fibrodisplasiaossificante progressiva. Um guia para famílias. International FOP Association (IFOPA) Winter Springs, Florida. Thirdedition.2009.
- DELAI, P. L. R. ; KANTANIE, S. ; SANTILI, C. ; KAPLAN, F. S. ; Fibrodisplasiaossificanteprogressiva: uma doença hereditária de interesse multidisciplinar. Revista Brasileira de Ortopedia – Vol.39, nº 5.2004.
- IFOPA InternationalFibrodysplasiaOssificans Progressiva Association. Acessado em 07 de junho de 2018. Acessado em 02 de abril de 2018.
- SERAFIM, P. H. et al. ; Achados radiológicos na fibrodisplasiaossificante progressiva. Revista Faculdade de Ciência Médica Sorocaba, v.7, n.4. P.23-25.2005.
- PINZAS, J. G. ; WONG, J. E. B. ; FERNÁNDEZ, M. A. P. ; ESPINOZA, M. A. R. ; Fibrodisplasiaossificante progressiva: diagnóstico em atenção primária. Revista Paul Pediátrica. 2013.
- KAPLAN, F. S. et al. ; Efficacy and safety of Palovarotene in fibrodysplasiaossificansprogressiva: a randomized, placebo-controlled, double-blind study. 2017.
- PALHARES, D. B. ; LEME, L. M. ; Miositeossificante progressiva: uma perspectiva no controle da doença. Jornal de Pediatria – Vol. 88, Nº5.2001.
- JÚNIOR, C. R. A. et al ;Fibrodisplasiaossificante progressiva: relato de caso e achados radiográficos. Radiologia Brasileira vol.38, nº1, p. 69-73.2005.
- EURORDIS RareDiseasesEurope. Acessado em 07 de junho de 2018.
- SHORE, E. M. ; KAPLAN, F. S.; Inherited human diseases of heterotopic bone formation. Rev. Rheumatol. 6, 518-527.2010.
- PAIM, L. B. et al. ;FibrodisplasiaOssificante Progressiva (FOP) em crianças: relato de três casos. Revista Brasileira de Reumatologia, v. 43, n. 2, p. 123-128.2003.
- FOP Brasil. Acessado em 16 de junho de 2018.
- FONSECA, J. E.et al ; Miositeossificante progressiva: Stone man. ACTA médica portuguesa ; 14:429-433.1999.
- DAHER, R. T. ; FARIA, R. S. ; MOREIRA, R. K. M. ; DAHER, R. T. ; Qual o seu diagnóstico? Whichisyourdiagnosis?. Radiologia Brasileira.2009.
- ABC Med.Acessado em 23 de junho de 2018.
- KAPLAN, F. S. et al ;Fibrodysplasiaossificansprogressiva. Best Pract Res ClinRheumatol. 22(1): 191-205.
- KAPLAN, F. S. ; PIGNOLO, R. J. ; MUKADDAM, M. M. A. ; SHORE, E. M. ; 26th Annual report of the fibrodysplasiaossificansprogressiva (FOP) collaborative research project. The Center for Research in FOP & Related Disordes.2017.
- KAPLAN, F. S. ; The skeleton in the closet. 528(1):7-11.2013.
- MENDES, R. A. B. et al ;Show de horrores: a ciência por trás das aberrações. Revista de Medicina e Saúde de Brasília. 5(2):333-58.2016.
- KAPLAN, F. S. et al ; Cuidados medicos de lafibrodisplasiaosificanteprogresiva: consideracionesactualesdeltratamento. El Consorcio Clínico Internacional enFibrodisplasiaOsificanteProgresiva. 1(2):1-81.2003.
- CAMPOS, D. M. et al ;Fibrodisplasiaossificante progressiva: relato de caso. Radiologia Brasileira. 38(5):393-395. 2005.
- CONNOR, J. M. ; EVANS, C. C. ; EVANS, D. A. P. ; Cardiopulmonary function in fibrodysplasiaossificans progressive. 36, 419-423.1981.
- RODRIGUES, D. C. ; Anquilose da articulação Temporomandibular. Universidade Federal de Minas Gerais.2011.
- ROMANI,F. ; KARAM, S. M. ; Fibrodisplasiaossificante progressiva: relato de caso. RevBras Ortop. 46(6)736-40.2011.
- RNTC – Rede Nacional de Terapia Celular. Acessado em 08 de julho de 2018.
- BONTRAGER, K. L. ; LAMPIGNANO, J. P. ; Tratado de posicionamento radiográfico e anatomia associada – 8. ed. 2015. Rio de Janeiro: Elsevier.
- GOMES,M. ; Guia de recursos. Associação Brasileira de FibrodisplasiaOssificante Progressiva – São Paulo.2017.